Schnyder Corneal Dystrophy
All content on Eyewiki is protected by copyright law and the Terms of Service. This content may not be reproduced, copied, or put into any artificial intelligence program, including large language and generative AI models, without permission from the Academy.

Disease Entity
Schnyder stromal dystrophy is a progressive bilateral corneal opacification that results from abnormal deposition of cholesterol and phospholipids in the cornea.
Genetics
It is a rare autosomal dominant stromal dystrophy, localized to the gene for UBIAD1-UbiA prenyltransferase domain containing 1, which is responsible for lipid metabolism.
History
First described by Van Went and Winbaut in 1924, and in 1929 a Swiss ophthalmologist named Schnyder published a report of a 3-generation family that all had this condition. It then became known as Schnyder crystalline corneal dystrophy.
Pathophysiology
The exact pathogenesis is unknown, but it is thought to result from a localized defect of the lipid metabolism. It is thought that there is an abnormal metabolism of phospholipid and cholesterol (particularly HDL) in the cornea. There has also been noted to be a loss of the corneal nerves, although frank neurotrophic keratopathy has not been documented.
Diagnosis
Slit lamp examination and testing corneal sensation, which is often decreased.
Corneal findings
- Corneal crystal: Bilateral cholesterol deposition in the anterior stroma. (Initial changes)
- Central corneal haze: Increased prevalence with age. (>40 years)
- Mid-peripheral haze: Increased prevalence with age. (>40 years)
- Arcus lipoides: With increasing age, arcus becomes prominent enough to be seen without the slit lamp. (23-39 years)
Symptoms
Increasing glare, light scatter, and resultant photopic vision loss can occur. Symptoms often worsen with age.
Systemic associations
- Genu valgum ("knocked knees")
- Hypercholesterolemia
Diagnostic procedures
- Slit lamp examination is the clue for diagnosis.
- Confocal microscopy in advanced stages may reveal the absence of corneal nerves.
- A recent observational study of 7 patients demonstrated small hyper-reflective deposits with increased background reflectivity.
- Anterior segment OCT has "shown diffuse high reflectivity in the epithelium, anterior, mid, and posterior stroma, which correspond to the hyperreflective deposits observed with IVCM and FF-OCT at the same level. Presence of epithelial hyperreflectivity was consistent with presence of areas of thick and irregular epithelium on epithelial mapping."
- Genetic testing looking for UBIAD1 mutations.
Differential diagnosis
- Fish eye disease.
- Tangier disease.
- Lecithin-cholesterol acyltransferase deficiency.
Treatment
No treatment is available to stop progression. Phototherapeutic keratectomy can remove subepithelial crystals if they are affecting visual acuity. Penetrating keratoplasty can be performed in eyes with advance disease but the disease can recur in the graft. One case series revealed that 54% of patients aged 50 years and older and 77% of patients aged 70 years and older had PKP.
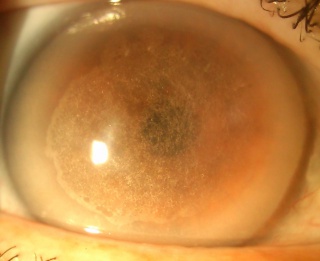
Elderly patient with advance distrophy. (Corneal crystals, midperipheral stromal haze and arcus lipoides)

References
- Weiss, J; Schnyder Corneal Distrophy. Curr Opin Ophthalmol 20: 292- 298
- Nowinska A, Wylegala E et al. Phenotype–Genotype Correlation in Patients With Schnyder Corneal Dystrophy.Cornea 2014;33:497–503
- Dolz-Marco R, Gallego-Pinazo R et al. Crystalline Subtype of Pre-Descemetic Corneal Dystrophy. J Ophthalmic Vis Res 2014; 9 (2): 269-271.
- Barchiesi B, Eckel R et al.The Cornea andDisorders of Lipid Metabolism.Surv Ophthalmol 36:1-22, 1991
- Weiss, J; Moller, H; The IC3D Classification of the Corneal Dystrophies. Cornea 2008;27(Suppl. 2):S1–S42
- Weiss JS. Visual morbidity in thirty-four families with Schnyder crystalline corneal dystrophy (an American Ophthalmological Society thesis). Trans Am Ophthalmol Soc 2007; 105:616-648.
- Ghazal W, Georgeon C, Grieve K, Bouheraoua N, Borderie V. Multimodal Imaging Features of Schnyder Corneal Dystrophy. J Ophthalmol. 2020 Mar 23;2020:6701816. doi: 10.1155/2020/6701816. PMID: 32280528; PMCID: PMC7125492.