Incontinentia Pigmenti
All content on Eyewiki is protected by copyright law and the Terms of Service. This content may not be reproduced, copied, or put into any artificial intelligence program, including large language and generative AI models, without permission from the Academy.
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, is a rare X-linked dominantly inherited syndrome manifesting at birth or early childhood. The cardinal feature is the appearance of characteristic progressive skin lesions, first presenting as vesiculobullous lesions and then progressing to whorl-like pigmentary lesions over four stages. IP also includes eye abnormalities, mostly occurring in the retina. The retinal findings stem from vascular occlusion and include neovascularization, hemorrhages, absence of foveal pit, and exudative and tractional retinal detachments. Pathologic changes in the central nervous system, teeth, and hair are also common in IP. Diagnosis is based on clinical systemic and ocular exam, as well as genetic testing. There is no treatment for the disease as a whole; however, symptoms may be managed by medical or surgical intervention.
Disease Entity
- ICD-10: Q82.3
- ICD-9: 757.33
- OMIM: 308300
Other names include: Bloch-Siemens incontinentia pigmenti, melanoblastosis cutis linearis, and pigmented dermatosis, Siemens-Bloch type.
IP was first described in the literature in 1906 by Garrod.
Epidemiology
The incidence of IP is 1 case per 40,000.[1] It is more common in Caucasians than other ethnicities. Because IP is an X-linked dominant disease, affected male fetuses usually do not survive, and therefore the great majority (97%) of living affected individuals are female.[2] [3] IP has high penetrance. Most persons with IP begin to express the phenotype within a few months of birth.
Pathophysiology
IP is due to mutation in the IKBKG gene (approved name: Inhibitor of the Kappa light polypeptide gene enhancer in B-cells, Kinase Gamma) which is located on the X chromosome at position q28.[3][4] This gene is also known as NEMO (Nuclear Factor κβ Essential Modulator) or IKK-γ (Inhibitor of nuclear factor Kappa-B Kinase, subunit gamma). This gene codes for one of three subunits of the IκB kinase enzyme complex which is part of the NF-κB cascade. NF-κB plays a critical role in regulating inflammation and the immune response to infection.[4][5][6] An approximately 11.7-kb deletion in the IKBKG gene that removes exons 4 through 10 is the cause of the majority of IP mutations.[5] This gene is involved in activating Nuclear Factor–κβ (NFκβ) which acts as a transcription factor, and is important in activating genes necessary for the production of cytokines and chemokines, as well as providing anti-apoptotic effects.[4][5][7]
With this mutated gene, endothelial cells and other cells throughout the body can over-express chemotactic factors specific for eosinophils, explaining why serum eosinophilia is common in IP patients. The epidermal skin lesions also show extensive involvement of eosinophils. It appears that the eosinophils, combined with other factors, lead to extensive inflammation, affecting not only the skin, but also endothelial cells. It is believed that this inflammation leads to vaso-occlusion, which results in ischemia, causing both the retinal and neurologic manifestations of IP.[8] However, to complicate matters, there is little evidence that the typical inflammatory pathology in the skin also occurs in the retina; furthermore, there is also a lack of evidence showing vaso-occlusion in the skin lesions.[9]
The ocular manifestations are the result of vaso-occlusions in the retinal arteries, resulting in subsequent ischemia. Avascular areas in the periphery of the retina develop secondary to this hypoperfusion and ischemia. As in many ischemic retinal conditions, neovascularization occurs with damaging sequela.[2][9]
Likewise, in the CNS (central nervous system), the cerebral and cerebellar atrophy as well as other neurologic sequela are believed to be due to vaso-occlusive ischemia and nerve cell death.[3][10] [11]
Risk Factors
Family history appears to be the only known risk factor for IP. Living males are likely to be affected with Klinefelter syndrome (47, XXY), where an extra X chromosome is present.[3]
Diagnosis
History
A thorough history of suspected IP patients will often yield a family history of the disease, as this is an X-linked dominantly inherited condition. There also may be a history of fetal demise in the family (more probable with male fetuses). Parents will often describe characteristic skin lesions (often described as a rash) starting within a few months of birth, which is the most common presenting complaint. Eye manifestations occur in 35-40% of patients and may cause a history of strabismus, leukocoria or visual problems. Neurologic problems occur in about 30% of patients with IP and may cause seizures, paralysis, motor and mental retardation, and developmental delays. A history of dental problems may be elicited in nearly 70% of affected individuals. Also, alopecia and nail abnormalities may be noted.[2][3]
Symptoms
Symptoms follow the main features of IP. The skin lesions themselves are fairly benign and typically only cause cosmetic issues, if any. Although the retina is often involved, this may not lead to symptomatic visual changes that could be detectable in an infant or young child on casual observation, necessitating ophthalmic examination. Cortical blindness may be present, causing total blindness or only light perception. Neurologic issues can cause significant symptoms; the most prominent are seizures, paralysis, and developmental delays. Other symptoms may include problems eating or transitioning to firm food, due to dental problems. Weak nails also may cause symptoms.
Physical Exam
A thorough dermatologic, neurologic, ophthalmologic, and dental exam should be performed.
Dermatologic Exam and Findings
In those suspected to have IP, a thorough skin examination must be done, although typical skin lesions are usually not difficult to find. The lesions go through four stages, beginning at or near birth and progressing to the final stage, which usually persists into adulthood (see images). These stages are:[3]
- Stage 1: Vesicular Stage - marked erythema with linear vesicles, bullae, and pustules; multiple crops may appear and are often present at birth or occur shortly after birth.
- Stage 2: Verrucous Stage - wart-like verrucous papules and keratotic patches; occurs in the first few weeks or months of life.
- Stage 3: Hyperpigmented Stage - swirling or whirling macular patches of hyperpigmentation; occurs anywhere between infancy and adolescence, but usually fade by early adulthood.
- Stage 4: Hypopigmented Stage - patchy areas of hypopigmentation, usually arranged in streaks or whorls; often with cutaneous atrophy; usually present by early adulthood.
These skin lesions have a specific pattern of distribution along the Lines of Blaschko, which are patterns of manifestation of dermal conditions reflected by the development of the skin during embryogenesis. These lesions most commonly occur on the side of the trunk but usually spare the face. Since these lesions occur so frequently in individuals with IP (nearly 100%), they are considered to be nearly pathognomonic if they fit the specific pattern, stage progression, and timeline. However, if these lesions are not present, IP cannot be ruled out.
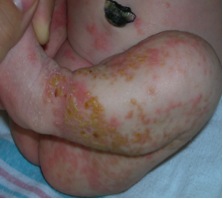
Stage 1 skin
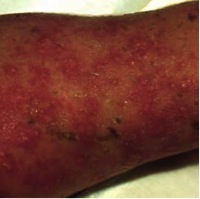
Stage 2 skin
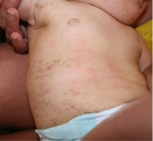
Stage 3 skin
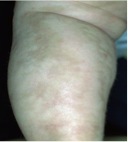
Stage 4 skin




(images used with permission from Dr. Jehad at www.Dermaamin.com)
Neurologic Exam and Findings
The neurological manifestations of IP are significant and lead to considerable morbidity.[3] Neurologic findings are present in 30% of IP cases. These abnormalities are typically limited to the brain and include convulsive disorders, spastic paralysis, motor retardation, and mental retardation. Seizures and developmental delays are the most common. Of note, significant CNS manifestations in the neonate with IP may denote a poor overall long-term prognosis. Importantly, one study suggested that over 90% of CNS symptoms will appear before age 2.[12] Brain imaging will often show cerebral ischemia, edema, and (later) atrophy as might be expected in the setting of vascular occlusion. Hydrocephalus has also been found in IP patients. If cortical blindness occurs, neurologic investigation is needed.
Ophthalmologic Exam and Findings
A thorough eye exam must be performed on those suspected of IP. Eye findings are seen in 35-40% of those with IP.[2][3] Both retinal and non-retinal findings are seen in IP. Retinal findings are common and include avascularity, neovascularization, hemorrhages, absence of a foveal pit, optic nerve atrophy, and exudative and retinal tractional detachments. Retinal detachment is the most commonly diagnosed eye finding in IP. Due to the vaso-occlusive nature of IP, a cherry red foveal spot may be seen as choroidal perfusion is not typically affected; however, observing the occlusive event in its acute stage is rare. A blunted foveal pit and absence of normal parafoveal vascular pattern are also characteristic of IP, caused by vessel occlusion and subsequent vascular remodeling.[2][9][13] Occlusions of retinal vessels in the macula and the periphery are typically seen and are often accompanied by diffuse mottled hypopigmentation. Retinal detachments, if present, nearly always occur within six years of age and are relatively rare in older patients. Additional non-retinal ocular manifestations include cataracts, uveitis, strabismus (18%), conjunctival pigmentation, optic atrophy (5%), and nystagmus.[2][9][14] Phthisis bulbi can be the result of ocular IP.
An avascular peripheral retina is a significant feature of IP’s retinal manifestations. Typically, the neovascularization of the retina is more pronounced in the periphery, similar to retinopathy of prematurity.[15] [16] Tufts of neovascularization can be found at the edges of the avascular retina. Fluorescein angiography is a critical study in the ophthalmic investigations for IP.[2] Often, enlargement of the foveal avascular zone is seen, which can be accompanied by large areas of avascularity anywhere in the retina.[2][9][15][16]
Cortical blindness may sometimes be present due to vascular occlusions in the occipital cortex in IP. Neurologic consultation and MRI brain imaging should be obtained to investigate this possibility.
Dental Exam and Findings
Dental problems occur in nearly 70% of affected individuals.[3] Even a limited dental exam will often yield missing, small, or abnormally shaped teeth. The general examination may also yield alopecia and nail abnormalities.
Clinical diagnosis
In 1993, Landy and Donnai proposed a set of clinical diagnostic criteria for IP.[17] The criteria focus on whether or not the suspected patient has a first-degree relative with IP. If this relative exists, then only one of the following is needed for diagnosis of IP is one of the following:
- History or evidence of typical skin lesions
- Pale, hairless, atrophic linear skin streaks
- Dental anomalies
- Retinal disease
- Multiple male miscarriages
If a first degree relative with IP cannot be established, then diagnostic criteria are separated into major and minor categories.
Major criteria: (skin lesions that occur in stages from infancy to adulthood)
- Erythematous lesions followed by vesicles anywhere on the body (sparing the face), usually in a linear distribution.
- Hyperpigmented streaks and whorls respecting Lines of Blaschko, occurring mainly on the trunk and sparing the face, fading in adolescence
- Pale, hairless, atrophic linear streaks or patches
Minor criteria:
- Hypodontia or anodontia (partial or complete absence of teeth), microdontia (small teeth), abnormally shaped teeth
- Alopecia, wiry coarse hair
- Mild nail ridging or pitting, hypertrophied, curved nails
- Retinal abnormalities
If a first-degree relative with IP cannot be established, one major criterion and two or more minor criteria are needed for diagnosis. If none of the minor criteria are present, then diagnosis other than IP should be considered.[17]
Genetic testing
The IKBKG gene (previously NEMO) is the only known gene associated with IP. Approximately 65% of affected individuals have an 11.7-kb deletion removing exons 4 – 10 from the IKBKG gene.[5] Targeted mutation analyses are available to look for this mutation. In addition to this deletion, other small IKBKG mutations have been found in those with IP, most often occurring in exon 10. Sequence analysis can detect these mutations. Also, in females with skewed lyonization in which the X chromosome with the mutant IKBKG allele is inactivated, X-chromosome inactivation studies may be useful. In suspected males, karyotyping and fluorescence in situ hybridization to look for evidence of 47, XXY may be done.[5]
Laboratory testing
Eosinophilia may occur (as high as 65% of leukocytes), particularly in stages I and II.[3]
Skin biopsy with histologic examination to evaluate for eosinophilic infiltration and/or extracellular melanin granules may be helpful in confirming the diagnosis in an individual with borderline or questionable findings in whom molecular genetic testing has not identified a disease-causing mutation.[11][13] Skin biopsy for diagnosis is rarely needed given the availability and sensitivity of genetic testing.
Differential diagnosis
The differential diagnosis for the retinal manifestations of IP include retinopathy of prematurity, familial exudative vitreoretinopathy, Eales retinopathy, sickle cell retinopathy, and the Norrie disease spectrum. However, skin findings are not associated with these conditions.
The differential diagnosis for the dermatologic findings in IP is large and can be divided based on the stage of the skin lesions. They include congenital herpetic infection, impetigo/epidermolysis bullosa, warts or molluscum contagiosum, hypomelanosis of Ito, scaring, and vitiligo.
Naegeli syndrome is a rare condition affecting the skin and ectoderm which can appear similar to the skin lesions of IP. It does not have significant eye findings.
Management
As IP has a genetic basis, at this time there is no cure and treatment is limited to treating symptoms.
General treatment
Because IP is a multi-faceted condition, dermatologic, genetic, ophthalmic, neurologic, and dental consultations should be obtained. Dermatologic consultation should include monitoring and evaluation for cutaneous squamous cell carcinoma in IP.[18] Generally, brain imaging should be obtained (MRI) to investigate the occlusive consequences of IP in the brain.[3][10] EEG should be obtained if seizures are present. Developmental therapy may be needed. Skin lesions should be managed symptomatically to avoid infection or excessive scarring.
Genetic therapies are not currently available for IP.
Ophthalmic treatment
The avascular retina should be treated similarly to retinopathy of prematurity with laser photocoagulation (or cryotherapy) of the avascular retina.[2][8][9][16] The occurrence of retinal detachments should be monitored with regular ophthalmological exams and surgical repair of detachments should be attempted by a vitreoretinal surgeon if appropriate. Vitrectomy with or without scleral buckle is commonly performed.
Intravitreal anti-vascular endothelial growth factor has been investigated.[19] While this may be a promising treatment, it has not become the standard of care.
Genetic Planning
If a proband is identified, clinical examination of family members and genetic testing of the mother is warranted.
If a female has IP (IKBKG mutation), the risk of conceiving a fetus with IP is 50%. However, most affected males die in utero, meaning that an affected mother has a near 33% probability to giving birth to either an affected female, an unaffected female, or unaffected male.[3]
In parents without IP or an IP-related IKBKG mutation who have had a child with IP, the risk of a subsequent child having IP is less than 1%.[3]
Prenatal testing
Prenatal diagnosis of fetuses with a family history of IP is possible via DNA analysis obtained by amniocentesis or chorionic villus sampling.
Additional Resources
- Incontinentia Pigmenti International Foundation
- Ectodermal Dysplasia Society
- NINDS Incontinentia Pigmenti Information Page
References
- ↑ Buinauskiene J, Buinauskaite E, Valiukeviciene S. Incontinentia pigmenti (bloch-sulzberger syndrome) in neonates. Medicina (Kaunas). 2005;41(6):496-9.
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 Goldberg MF, Custis PH. Retinal and other manifestations of incontinentia pigmenti (bloch-sulzberger syndrome). Ophthalmology. 1993 Nov;100(11):1645-54.
- ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 3.11 3.12 Scheuerle A, Ursini MV. Incontinentia pigmenti. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews. Seattle (WA): University of Washington, Seattle. All rights reserved; 1993.
- ↑ 4.0 4.1 4.2 Nelson DL. NEMO, NFkappaB signaling and incontinentia pigmenti. Curr Opin Genet Dev. 2006 Jun;16(3):282-8.
- ↑ 5.0 5.1 5.2 5.3 5.4 Fusco F, Pescatore A, Bal E, Ghoul A, Paciolla M, Lioi MB, et al. Alterations of the IKBKG locus and diseases: An update and a report of 13 novel mutations. Hum Mutat. 2008 May;29(5):595-604.
- ↑ Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci. 2003 Oct;6(10):1072-8.
- ↑ Gilmore TD. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene. 2006 Oct 30;25(51):6680-4.
- ↑ 8.0 8.1 Goldberg MF. The skin is not the predominant problem in incontinentia pigmenti. Arch Dermatol. 2004 Jun;140(6):748-50.
- ↑ 9.0 9.1 9.2 9.3 9.4 9.5 Goldberg MF. The blinding mechanisms of incontinentia pigmenti. Ophthalmic Genet. 1994 Jun;15(2):69-76.
- ↑ 10.0 10.1 Wolf NI, Kramer N, Harting I, Seitz A, Ebinger F, Poschl J, et al. Diffuse cortical necrosis in a neonate with incontinentia pigmenti and an encephalitis-like presentation. AJNR Am J Neuroradiol. 2005 Jun-Jul;26(6):1580-2.
- ↑ 11.0 11.1 Ehrenreich M, Tarlow MM, Godlewska-Janusz E, Schwartz RA. Incontinentia pigmenti (bloch-sulzberger syndrome): A systemic disorder. Cutis. 2007 May;79(5):355-62.
- ↑ Pinheiro A, Mathew MC, Thomas M, Jacob M, Srivastava VM, Cherian R, et al. The clinical profile of children in india with pigmentary anomalies along the lines of blaschko and central nervous system manifestations. Pediatr Dermatol. 2007 Jan-Feb;24(1):11-7.
- ↑ 13.0 13.1 Berlin AL, Paller AS, Chan LS. Incontinentia pigmenti: A review and update on the molecular basis of pathophysiology. J Am Acad Dermatol. 2002 Aug;47(2):169,87; quiz 188-90.
- ↑ Minic S, Obradovic M, Kovacevic I, Trpinac D. Ocular anomalies in incontinentia pigmenti: Literature review and meta-analysis. Srp Arh Celok Lek. 2010 Jul-Aug;138(7-8):408-13.
- ↑ 15.0 15.1 Nix RR, Apple DJ. Proliferative retinopathy associated with incontinentia pigmenti. Retina. 1981;1(3):156-61.
- ↑ 16.0 16.1 16.2 Watzke RC, Stevens TS, Carney RG,Jr. Retinal vascular changes of incontinentia pigmenti. Arch Ophthalmol. 1976 May;94(5):743-6.
- ↑ 17.0 17.1 Landy SJ, Donnai D. Incontinentia pigmenti (bloch-sulzberger syndrome). J Med Genet. 1993 Jan;30(1):53-9.
- ↑ King A, Liy-Wong C, Bose R. Cutaneous squamous cell carcinoma as a late and underrecognized manifestation of incontinentia pigmenti: A systematic review, JAAD Reviews. 2024;1:S93-S96. https://doi.org/10.1016/j.jdrv.2024.07.005.
- ↑ Cernichiaro-Espinosa LA, Patel NA, Bauer MS, Negron CI, Fallas B, Pogrebniak A, Berrocal AM. Revascularization After Intravitreal Bevacizumab and Laser Therapy of Bilateral Retinal Vascular Occlusions in Incontinentia Pigmenti (Bloch-Sulzberger Syndrome). Ophthalmic Surg Lasers Imaging Retina. 2019 Feb 1;50(2):e33-e37.
Authors: Peter A. Karth, MD; Andrew Johnson, JMS; Dennis P. Han, MD