Fibrous Dysplasia
All content on Eyewiki is protected by copyright law and the Terms of Service. This content may not be reproduced, copied, or put into any artificial intelligence program, including large language and generative AI models, without permission from the Academy.
Disease Entity
Fibrous dysplasia is a benign disorder of bone. It can involve any bone, but most commonly affects the long bones of the extremities or the craniofacial skeleton. There are three forms of fibrous dysplasia: monostotic (involving a single skeletal site), polyostotic (multiple sites), and the McCune Albright Syndrome (polyostotic fibrous dysplasia with endocrine and skin changes). In all three forms, normal bone is replaced by fibrous-osseous tissue.
Disease
- Fibrous dysplasia monostotic (ICD-9 # 733.29)
- Polyostotic fibrous dysplasia (ICD-9 # 756.54)
- Osteodystrophy NEC (ICD-9 756.59)
- McCune-Albright syndrome (ICD-9 #756.59)
Etiology
Fibrous dysplasia occurs due to an activating somatic missense mutation of a G protein that interferes with normal bone maturation and resorption. [1]
Risk Factors
An equal distribution is noted between sexes in monostotic and polyostotic fibrous dysplasia, and the disease can be found among all racial groups. [2][3][4] McCune-Albright Syndrome may be seen more commonly in female patients. [5][1]
General Pathology
Monostotic fibrous dysplasia is the most frequent form, and accounts for approximately 75% of all cases. [6][7][5] The remaining 20-30% are of the polyostotic variant, which tends to produce more severe bone deformity. [5][8]
A small subset of polyostotic fibrous dysplasia associated with endocrine dysfunction and characteristic café-au-lait skin pigmentation is known as the McCune-Albright Syndrome. This syndrome accounts for approximately 3% of polyostotic disease. [9] Precious puberty is the most frequent hyperfunctioning endocrinopathy; others include acromegaly, hyperthyroidism, diabetes, hyperparatuitarism, hypercalcemia, and adrenal disorders. [10][6]
The most common sites of involvement in monostotic fibrous dysplasia are the craniofacial skeleton, ribcage, femurs and tibias. [2] By contrast, in polyostotic disease, dysplasia most often occurs in the femurs and tibias, followed by the craniofacial skeleton, pelvis, ribs, humeri, and spine. [2][8][6] No evidence supports the progression of monostotic to polyostotic dysplasia.
Twenty to thirty percent of all fibrous dysplasia involves the craniofacial skeleton. Commonly affected sites include the maxilla, ethmoid, sphenoid and frontal bones. [7][11][5][4] Craniofacial dysplasia may be seen in over half of polyostotic patients and 10-35% of monostotics. [12][5][2] Fibrous dysplasia of multiple adjacent craniofacial bones is considered monostotic disease. [1][11]
Primary bone tumors of the orbit represent a small percentage of all orbital tumors (0.6-2%). [4] Fibrous dysplasia represents approximately 18% of all primary orbital bone tumors. [4] The majority of cases with orbital involvement have monostotic fibrous dysplasia of the frontal, sphenoid or ethmoid bones. [4]
Pathophysiology
Fibrous dysplasia is thought to result from an activating somatic missense mutation of the GNAS1 gene on chromosome 20. The protein transcript of GNAS1 is a stimulatory signaling G protein, Gs-alpha, and the mutated protein causes prolonged adenylyl cyclase activity and overproduction of intracellular cyclic adenoside monophosphate, (cAMP). [13][14][10][6][4][1][2]. More recent studies have also implicated the WNT/ β-catenin signaling pathway. [6]
This mutation triggers the replacement of normal bone by immature bone marrow stromal cells in a fibrous matrix. An arrest of typical bone maturation leads to abnormal, functionally impaired osteoblasts that produce spicules of poorly organized bone. [14][13][7][3]Increased levels of IL-6 stimulate osteoclastic bone resorption at fibrous dysplasia sites, resulting in poor mineralization. [3][9]
Primary prevention
No known prevention exists for fibrous dysplasia.
Diagnosis
The diagnosis of fibrous dysplasia can often be made based on the clinical and radiographic appearance, but in some cases may be confirmed with a tissue biopsy.
History
Monostotic fibrous dysplasia patients present symptomatically between age ten to thirty, on average, whereas the majority of polyostotic patients present prior to the age of ten.
The natural course of fibrous dysplasia is a matter of some debate. Historically, it was often regarded as a disease of childhood that tended to become quiescent following puberty. Multiple studies, however, have reported both active disease and progression in adults. [15][11][13][10]
Physical examination
The exam will vary based on the site of involvement and degree of dysplasia. Patients with craniofacial FD should receive a full ophthalmologic exam, including detailed external exam with attention to any facial asymmetry, pupillary function, extraocular movements, visual fields, exophthalmometry, visual acuity, color vision, intraocular pressure measurement, slit-lamp and fundus exam. Decreased visual acuity may not present until late in the course of optic neuropathy associated with fibrous dysplasia.
Signs and Symptoms
The most common presenting symptoms of fibrous dysplasia include pain, swelling and disfigurement due to fibroblastic expansion of the affected sites. Within the craniofacial skeleton, this can manifest as headaches, proptosis, and facial asymmetry. [12][11][16]. Visual impairment and hearing loss are the most common reported neurologic complications of fibrous dysplasia. [12][13][11][16] In addition to vision changes and globe displacement, bony thickening of the skull, its bony canals and foramena, and adjacent sinuses may also produce sinus collapse, nasal obstruction, extraocular muscle palsies, trigeminal neuralgia, anomia, and epiphora. [17][18]
Clinical diagnosis
Clinical diagnosis relies on both facial changes and imaging characteristics. Symptoms can include long standing facial asymmetry, proptosis, orbital pain, diffuse headache, double vision, visual loss, hearing loss, malocclusion, cranial nerve palsies, or tearing. [4] Careful attention to facial symmetry, cranial nerve function, visual acuity, visual fields, external ocular adnexal exam as well as orbital evaluation helps to support the diagnosis. Skin should be checked for café-au-lait spots (seen in McCune-Albright Syndrome)[19][20].
Diagnostic procedures
Fibrous dysplasia was initially studied using plain X-rays, and its characteristic mottled, sclerotic appearance described as resembling “ground-glass.” Today, lesions may be well characterized on computed tomography. The radiographic features vary based on the proportion of normal, mineralized bone to fibrous tissue in any given lesion. Early in the disease course, cystic and sclerotic areas are noted. As dysplasia progresses, a heterogeneous “pagetoid” pattern of radiolucent and radiopaque areas is seen, reflecting regions of fibrous stroma and opaque, osseous changes, respectively. [11][4] Typically, fibrous dysplasia has smooth cortical margins and no soft tissue involvement. [3][11][2] On MRI, lesions are low to isointense on T1 and T2-weighted images, and demonstrate moderate enhancement with gadolinium. [5][4][1]
Radiographic optic nerve bony encasement is exceedingly common in craniofacial fibrous dysplasia, and may approach rates as high as 50-90% of patients. [14][21] The relationship between optic canal stenosis and visual compromise is uncertain, however, as large studies have demonstrated that the majority of patients with completely encased nerves remain asymptomatic without evidence of optic neuropathy. (Figures 1 and 2) [10][14][16]
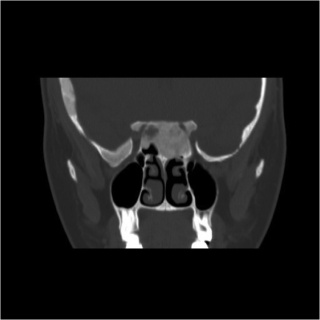
Figure 1. Bony optic nerve encasement in craniofacial fibrous dysplasia.
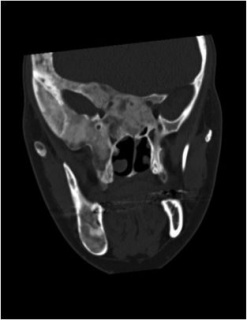
Figure 2. Craniofacial fibrous dysplasia.


Firm, gritty, pink or white tissue is seen on gross sectioning. [4][2]. Histologically, dysplastic bone has a classic appearance of dense fibrous stroma of low to moderate cellularity surrounding irregularly shaped bony trabeculae. This pattern has been described as resembling “Chinese characters.” (Figure 3) [6][4] No osteoblastic rimming of trabeculae is seen, which is of note in distinguishing fibrous dysplasia from other malignant entities, such as ossifying fibroma. [4][6][1] The surrounding stroma may contain aneurysmal bone cysts, cartilaginous nodules, and myxomatous areas. [4]

Figure 3. Dense fibrous stroma with irregular bony trabeculae. Courtesy of Riddle et al.
Laboratory test
Serum alkaline phosphatase may be elevated in up to one third of FD patients, regardless of disease severity or the presence of fractures. [1]
Differential diagnosis
Fibrous dysplasia can resemble other entities including meningioma, Paget’s disease or other osteodystrophies of the skull base, eosinophilic granuloma, Hand-Schuller-Christian disease, and low-grade central osteosarcoma. [17][3][5] Unlike fibrous dysplasia, meningiomas will exhibit a homogenous, sclerotic appearance if hyperostosis is present (Figure 4), may involve surrounding soft tissue, and display rapid contrast enhancement on MRI. [11]

Figure 4. Meningioma initially diagnosed as fibrous dysplasia.
Management
The treatment of fibrous dysplasia is guided by the severity of the disease and its complications, and options include careful observation and surgical intervention. No proven therapy is currently known for halting disease progression or preventing malignant transformation. The disease seems to stablilze with skeletal maturity. [1]
Medical therapy
Patients with minimal or mild symptoms may be observed with periodic CT or MRI imaging, and encouraged to maintain bone density through diet and exercise. [5][6]. Bisphosphonates have been utilized in efforts to halt bone resorption, and some studies suggest some improvement in pain, biochemical markers of turnover, and radiographic lesions. [22]
Medical follow up
Visual impairment in fibrous dysplasia may be acute or chronic in nature. Multiple mechanisms for its development have been advanced, including progressive canal stenosis, compression by an external mass, optic nerve traction secondary to displacement of the globe, and rarely, vascular compromise of the optic nerve. [23][16] Visual loss attributed to progressive canal narrowing tends to follow a chronic, gradual course. Many case reports describe acute vision loss due to external compression of the optic nerve by mucoceles, hemorrhage into an area of dysplastic bone, and aneurysmal bone cysts. [18][24][11][16][21]
Surgery
Surgical intervention remains the mainstay of treatment for symptomatic fibrous dysplasia, and may be undertaken in the setting of severe disfigurement, pathologic fractures, and other sequelae, including vision loss.
In patients with evidence of visual impairment and optic nerve encasement on imaging, therapeutic optic nerve decompression is advocated in efforts to preserve visual function. Less consensus exists regarding the treatment of asymptomatic patients with radiographic optic nerve encasement. Many experts recommend expectant management with careful follow-up as most individuals will retain full visual function. [10][14][21]. Others, however, support the practice of prophylactic optic nerve decompression given the risk of irreversible visual loss. [24][7].
Studies examining recurrence after surgical removal have found that residual tissue exhibits relatively normal growth rates, and that increased rates of malignancy are not seen following partial removal of dysplastic bone. [7]
Complications
Visual impairment and hearing loss are the most common reported neurologic complications of fibrous dysplasia. [12][13][11][16]
Malignant degeneration of fibrous dysplasia has been reported to occur in 0.5-1% of cases [25][16]. This risk is increased in polyostotic disease, particularly in McCune-Albright patients, and following radiation [8][7]. Transformation more frequently occurs in lesions within the craniofacial bones and femur. [8] The most common malignancy is osteogenic sarcoma, followed by fibrosarcoma, chondrosarcoma, and malignant fibrous histiocytoma. [7][6] The mean time interval from diagnosis to malignancy is 13.5 years, and clinical signs include rapid increase in lesion size or pain, intralesional necrosis or bleeding, and elevation of serum alkaline phosphatase levels. [8][7].
Prognosis
Fibrous dysplasia has a good prognosis with low rates of malignant transformation. The disease may stabilize with bone maturation. [1]
Additional Resources
References
- ↑ Jump up to: 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 WHO Classification of Tumours “Pathology and Genetics of Head and Neck Tumours,” 2005, 321-322, 336.
- ↑ Jump up to: 2.0 2.1 2.2 2.3 2.4 2.5 2.6 WHO Classification of Tumours “Pathology and Genetics of Tumours of Soft Tissue and Bone,” 2002. 341-342, 357-359.
- ↑ Jump up to: 3.0 3.1 3.2 3.3 3.4 Feller L, Wood N, Khammissa R, Lemmer J, Raubenheimer E. The nature of fibrous dysplasia. Head Face Med 5:22, 2009.
- ↑ Jump up to: 4.00 4.01 4.02 4.03 4.04 4.05 4.06 4.07 4.08 4.09 4.10 4.11 4.12 Selva D, White V, O’Connel J, Rootman J. “Primary Bone Tumors of the Orbit.” Survey of Ophthalmology 49(3):330-331, May-June 2004.
- ↑ Jump up to: 5.0 5.1 5.2 5.3 5.4 5.5 5.6 5.7 Lustig LR, Holliday MJ, McCarthy EF, Nager GT. Fibrous dysplasia involving the skull base and temporal bone. Arch Otolaryngol Head Neck Surg. 127:1239–1247, 2001.
- ↑ Jump up to: 6.0 6.1 6.2 6.3 6.4 6.5 6.6 6.7 6.8 Riddle N, Bui M. Fibrous Dysplasia. Archives of Pathology & Laboratory Medicine 137 (1):134-138, January 2013.
- ↑ Jump up to: 7.0 7.1 7.2 7.3 7.4 7.5 7.6 7.7 Papay FA, Morales L Jr, Flaharty P. Optic nerve decompression in cranial base fibrous dysplasia. J Craniofac Surg 6:5–14, 1995.
- ↑ Jump up to: 8.0 8.1 8.2 8.3 8.4 Parekh SG, Donthineni-Rao R, Ricchetti E, Lackman RD. Fibrous Dysplasia. J Am Acad Orthop Surg. 12(5):305-13, Sep-Oct 2004.
- ↑ Jump up to: 9.0 9.1 Chapurlat RD, Orcel P. Fibrous dysplasia of bone and McCune-Albright syndrome. Best Pract Res Clin Rheumatoly 22(1):55-69, March 2008.
- ↑ Jump up to: 10.0 10.1 10.2 10.3 10.4 Cutler CM, Lee JS, Butman JA, FitzGibbon E, Kelly MH, Brillante BA, Feuillan P, Robey PG, DuFresne CR, Collins MT. Long-term outcome of optic nerve encasement and optic nerve decompression in patients with fibrous dysplasia: risk factors for blindness and safety of observation. Neurosurgery 59(5):1011-7, 2006.
- ↑ Jump up to: 11.0 11.1 11.2 11.3 11.4 11.5 11.6 11.7 11.8 11.9 Katz BJ, Nerad JA. Ophthalmic manifestations of fibrous dysplasia: a disease of children and adults. Ophthalmology 105:2207–2215, 1998.
- ↑ Jump up to: 12.0 12.1 12.2 12.3 Sassin JF, Rosenberg RN. Neurological complications of fibrous dysplasia of the skull. Arch Neurol 18:363–369, 1968.
- ↑ Jump up to: 13.0 13.1 13.2 13.3 13.4 Lee J, FitzGibbon E, Chen YR, Kim HJ, Lustig LR, Akintoye SO, Collins MT, Kaban LB. Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet J Rare Dis. 7(Suppl 1): S2, 2012.
- ↑ Jump up to: 14.0 14.1 14.2 14.3 14.4 Amit M, Collins M, FitzGibbon E, Butman J, Fliss D, Gil Z. Surgery versus Watchful Waiting in Patients with Craniofacial Fibrous Dysplasia – a Meta-Analysis. PLoS One 6(9): e25179, 2002.
- ↑ Davies ML, Macpherson P. Fibrous dysplasia of the skull: disease activity in relation to age. Br J Radiol 64(763):576–579, 1991.
- ↑ Jump up to: 16.0 16.1 16.2 16.3 16.4 16.5 16.6 Michael C, Lee A, Patrinely J, Stal S, Blacklock JB. Visual loss associated with fibrous dysplasia of the anterior skull base: Case report and review of the literature. Journal of Neurosurgery, 92(2):350-354, 2000.
- ↑ Jump up to: 17.0 17.1 Gass, JD. Orbital and Ocular Involvement in Fibrous Dysplasia. Southern Medical Journal 58:324-9, Mar 1965.
- ↑ Jump up to: 18.0 18.1 Liakos GM, Walker CB, Carruth JAS. Ocular complications in craniofacial fibrous dysplasia. Br J Ophthalmol 63:611–616, 1979.
- ↑ McCune DJ. Osteitis fibrosa cystica: The case of a nine year old girl who also exhibits precocious puberty, multiple pigmentation of the skin and hyperthyroidism. Am J Dis Child 52:743, 1937.
- ↑ Albright F, Butler AM, Hampton AO, Smith P. Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation and endocrine dysfunction, with precocious puberty in females: report of five cases. New Eng. J. Med. 216 (17): 727–746, 1937.
- ↑ Jump up to: 21.0 21.1 21.2 Lee JS, FitzGibbon E, Butman JA, Dufresne CR, Kushner H, Wientroub S, Robey PG, Collins MT. Normal vision despite narrowing of the optic canal in fibrous dysplasia. N Engl J Med 347:1670-6, 2002.
- ↑ Chapurlat RD, Hugueny P, Delmas P, Meunier P. Treatment of fibrous dysplasia of bone with intravenous pamidronate: long-term effectiveness and evaluation of predictors of response to treatment. Bone 35(1):235-242, July 2004.
- ↑ Seiff SR. Optic nerve decompression in fibrous dysplasia: indications, efficacy, and safety. Plast Reconstr Surg. 100(6):1611-2, November 1997.
- ↑ Jump up to: 24.0 24.1 Chen YR, Breidahl A, Chang CN. Optic nerve decompression in fibrous dysplasia: indications, efficacy, and safety. Plast Reconstr Surg 99:22–30, 1997.
- ↑ Edgerton MT, Persing JA, Jane JA. The Surgical Treatment of Fibrous Dysplasia with Emphasis on Recent Contributions from Cranio-Maxillo-Facial Surgery. Ann Surg 202:459–479, 1985.